Mitochondrial Homeostasis and Brain Development:
The brain is a highly energy-demanding organ that consumes more than 20% of the total energy produced by the organism. Most of the energy produced in the form of ATP is produced by mitochondria. A hallmark of brain metabolism is the tight coupling between energy demand and supply, which involves constant regulation of mitochondrial homeostasis and correct localization of mitochondria in the distinct sub-cellular compartments of the polarized neurons to address their unique regional metabolic needs in response to signaling cues. Mitochondrial number and health are under the control of three pathways: biogenesis, fusion-fission cycles, and mitochondrial quality control. Mitochondrial biogenesis generates new mitochondria via replication of mtDNA. Mitochondrial fusion allows exchanging matrix content and mtDNA molecules between mitochondria, which favors optimal mitochondrial physiology by diluting mutated MtDNA and rescuing damaged mitochondria via acquisition of key components from healthy mitochondria. The mitochondrial quality control system is essential to maintain a functional mitochondrial population in neurons, which occurs via mitophagy to clear aged and dysfunctional mitochondria.
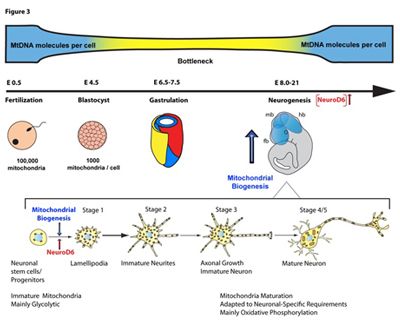
The Chiaramello laboratory focuses on the regulation of mitochondrial biogenesis in developing neurons, a complex process that requires a coordinated mitochondrial-nuclear crosstalk. Our studies have demonstrated a link between the neurogenic basic Helix-Loop-Helix (bHLH) transcription factor NeuroD6 during the early stages of neuronal differentiation. NeuroD6, which contributes to the specification of multipotential progenitors towards a glutamatergic pyramidal fate during corticogenesis, links neuritogenesis to mitochondrial biogenesis via replication of mtDNA. NeuroD6 also promotes an OXPHOS metabolism at the outset of neuronal differentiation, resulting in increased ATP production and generating a bioenergetic reserve. NeuroD6 also confers cellular tolerance to mitochondrial stressors and oxidative stress, known to compromise neurogenesis and cause specific neurodevelopmental disorders and neurodegenerative diseases.
Inherited Mitochondrial Diseases:
The Chiaramello lab studies a group of rare mitochondrial diseases defined by insufficient ATP levels due to impaired oxidative phosphorylation (OXPHOS). The OXPHOS system is formed of a series of multi-subunit enzymes embedded in the inner mitochondrial membrane responsible to make ATP upon electron transfer. Thus, these diseases are referred to as mitochondrial respiratory disorders (MRDs). MRD patients harbor inherited mutation that map un their mitochondrial or nuclear genome thereby impairing OXPHOS activity and ATP synthesis. Currently, no therapeutic options are available to the patients, resulting in significant disability and a constellation of multi-system clinical symptoms.
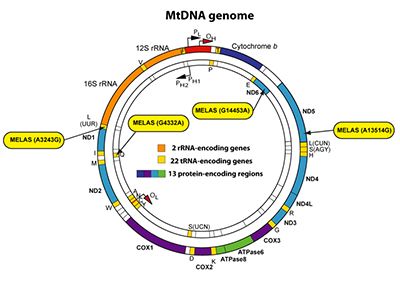
In collaboration with Dr. Andrea Gropman, a pediatric neurologist specialized in mitochondrial disorders, we study the mitochondrial disease MELAS (Mitochondrial Encephalopathy with Lactic Acidosis and Stroke-like episodes). MELAS is the most common mitochondrial respiratory disorder that is a childhood-onset progressive neurodegenerative disease. MELAS is due to maternally inherited mutations in the mitochondrial genome. Most MELAS patients exhibit an A to G substitution at position 3243 of the mitochondrial gene for tRNALEU(UUR), which only affects a subset of the multi-copy mitochondrial genome, causing heteroplasmy. Thus, MELAS patients harbor a mixed population of functional and dysfunctional mitochondria. Dysfunctional mitochondria carry a higher load of mutant mitochondrial DNA molecules than functional mitochondria. As a result, MELAS pathogenesis is driven by a chronic state of energy deficit due to the inability of dysfunctional mitochondria to generate sufficient ATP levels via OXPHOS.
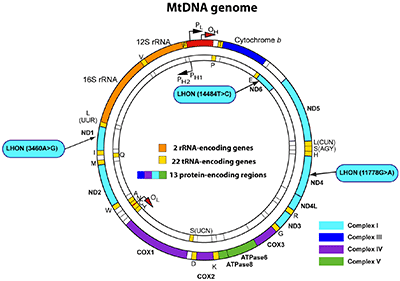
Together with Dr. Gropman and Lisa Thompson, a patient advocate for the LHON-Plus disease, we conduct patient-based research on the maternally inherited mitochondrial disease Leber’s hereditary optic neuropathy associated with the multiple-like illness, also referred to as LHON-Plus. We aim to understand the molecular pathogenic mechanisms responsible for mitochondrial dysfunction. The most prevalent mitochondrial variants, m.11778G>A, m.3460A>G and m.14484T>C, result in Complex I deficiency, leading to chronic energy deficiency affecting various organs with high energy demand. This multi-organ dysfunction results in a high clinical phenotypic variability among patients, making the clinical diagnosis challenging. Currently, patients only have access to palliative therapies, as therapeutic options for LHON-Plus remain elusive. We aim to explore a novel therapeutic approach to mitigate the mitochondrial dysfunction in patients with LHON-Plus. Our research is funded by the Department of Defense.
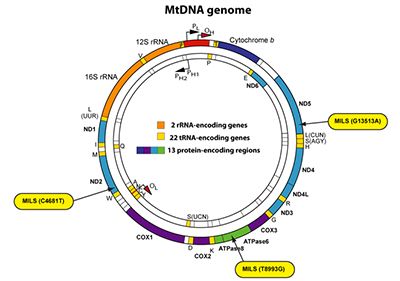
In addition, our team is also engaged in the study of another mitochondrial disease, called MILS (Maternally Inherited Leigh Syndrome), which is an infantile subacute necrotizing encephalopathy caused by mutations in the nuclear or mitochondrial genome affecting the OXPHOS system. Several mitochondrial mutations can cause MILS, with all of them affecting one OXPHOS complex, thereby reducing the production of mitochondrial ATP. Patients with more than 90% of heteroplasmy are classified as having MILS, whereas patients with a lower heteroplasmy exhibit symptoms of NARP (Neuropathy, Ataxia and Retinitis Pigmentosa). Both MILS and NARP are multisystem disorders that are maternally inherited.
Small molecule and epigenetic-based therapeutic approaches
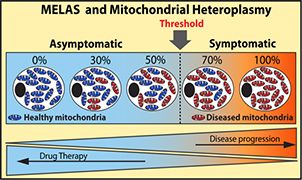
One promising strategy is to reduce the load of diseased or dysfunctional mitochondria to a sub-threshold. One of our main therapeutic avenues is to efficiently boost mitochondrial biogenesis via pharmacological means to compensate for the OXPHOS deficit associated with mitochondrial respiratory disorders. Our objective is to boost biogenesis of functional mitochondrial mass. This shift in heteroplasmy would optimize OXPHOS functions and alleviate symptomatic manifestations and mitigate the chronic energy deficit in patients.
